
Corrosion is the breakdown of materials due to chemical reactions. It is usually oxidation with air molecules, often occurring in the presence of water. Corrosion also occurs when an acidic/basic corrosive substance comes into contact with another material.
Steel and Concrete have become the most common materials for artificial structures over the last hundred or so years, with the use of the composite material, Concrete reinforced with steel, becoming one of the most popular methods for civil construction.
The historical reasons for steel-reinforced Concrete’s popularity are not hard to find: its cheapness, high structural strength, mouldability, fire resistance, and supposed imperviousness to the external environment, while requiring little or no maintenance, provide a virtually unbeatable combination.
To harness these properties, both national and international standards have been developed. The standards for Concrete and steel were initially defined principally by compositional limits and strength, which has continued to be the primary quality control.
Until the 1950s, it was assumed that when steel was encased in the alkaline Concrete matrix, neither would suffer from any degradation for the indefinite future.
However, evidence of degradation was noted as early as 1907 (Knudsen, 1907), where it was observed that the addition of chlorides to Concrete could allow sufficient corrosion of the steel to cause cracking of the Concrete.
The implicit assumption by many civil engineers that reinforced Concrete’s virtually infinite durability is valid has proven to be accurate in several cases, with structures reaching their design lives without any evidence of structural degradation. However, it is now evident that in areas with an aggressive atmosphere, the Concrete can be damaged, or the steel can corrode, dramatically shortening the specified design life.
Also Read: What is Civil Engineering? | History and Functions
The current design life for UK highways was initially set at 120 years. Despite all the evidence from highway structures showing significant problems after a short period, the Figure remains extremely optimistic, even though no corrosion design-life analysis is required.
This head-in-the-sand approach can be contrasted with the reality illustrated by research (Bamforth, 1994), which shows that the estimated time to corrosion activation of steel reinforcement in modern Concrete with the designated cover can be as low as 5.5 years at a 0.4% chloride level.
These research findings are in good accordance with site investigations. A substantial number of structures have been found to have their steel reinforcement sufficiently corroded within 20 years of construction, rendering them structurally unsound.
Even after the publicity surrounding the large number of structures exhibiting acute signs of distress 25 or so years into a designed 120-year life span, there is still a body of engineers who believe that all that is required to achieve any specified design life in a hostile environment is to provide a higher Concrete grade with the same design and maintenance of the structure.
This contention does not align with the facts, meaning that publications like this book will address past civil engineering miscalculations and those that may be committed in the future.
The traditional use of cathodic protection is to prevent corrosion of steel objects in the ground or water, which remains its most common application. It is almost universally adopted on ships, oil rigs, and oil and gas pipelines. Over the last 50 years, cathodic protection has evolved from being a black art to a science for these applications.
Over the past 30 or so years, there has been a steady increase in the use of cathodic protection for rehabilitating reinforced Concrete structures exhibiting signs of distress.
The most common damage mechanism is chloride-induced corrosion of the steel reinforcement, which is typically what cathodic protection systems are designed to prevent.
Initially, the cathodic protection techniques for reinforced Concrete followed the practice of ‘traditional’ impressed current systems closely, but, particularly over the past decade or so, there have been significant developments that have allowed the protection of Concrete structures to become a legitimate and yet distinctly different part of the cathodic protection mainstream with its protection criteria, anode types, and even power supplies.
The object of this volume is to introduce the current state of the art in the cathodic protection of Concrete and outline other related electrochemical techniques for stopping the corrosion of steel reinforcement.
Some guidelines on what cathodic protection is and how and when to use it are also discussed, so that a practising civil engineer or owner should have an introduction to the murky world of cathodic protection for reinforced Concrete.
Recommended Articles
- How To Calculate The Number Of Blocks In a Wall
- How To Simply Estimate The Quantity of Plastering Materials
- Quantities Of Materials For Concrete: The Simple Calculation You Need To Know
- The Basic Civil Engineering Materials: What You Need To Know
- Everything You Need To Know About Ready-Mix Concrete (RMC)
- Building Information Modelling for Engineers: The Ultimate Guide
- Key Skills Every Civil Engineer Need To Learn In 2026
- Best Structural Analysis Software for Civil Engineers In 2026
Also Read: Best Types of Kitchen | All You Need To Know- Right Now
Electrochemical Corrosion
Electrochemical reactions are widely used in industry for processes such as anodising and chloride production. They are used directly by most people every day of their lives when using a battery.
A surprising number of engineers vaguely remember an explanation in chemistry classes of how a battery operates. This is commonly reiterated as being about electrolytes, with ions swimming about, with anodes and cathodes making an appearance, and then dismissed as unnecessary in ‘proper’ civil or mechanical engineering.
Unfortunately for those who do not like electrical circuits, corrosion is also an electrochemical process and is of great economic importance, as those with old cars will testify, and has been estimated to consume 4% of the Gross National Product of, for example, the United States (Bennett et al., 1978). This percentage is likely to be of the same order globally.
In all low-temperature corrosion reactions and the processes given above, an electrochemical cell is required for them to occur. This cell comprises an anode and a cathode separated by an electrolytic conductor (electrolyte) with a metallic connection.
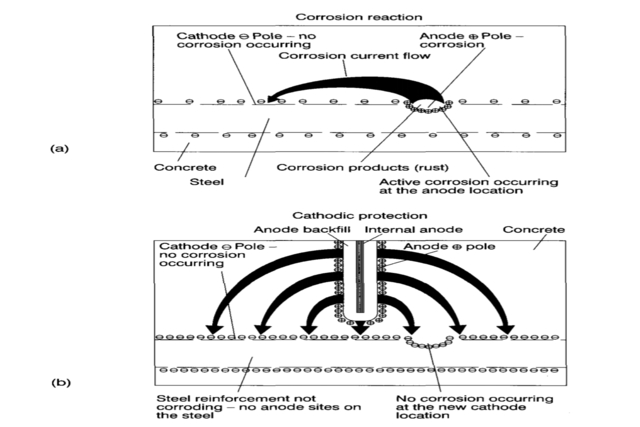
This is shown schematically below. A practical definition of an anode is the area where corrosion occurs, while the cathode is the area where no corrosion occurs.
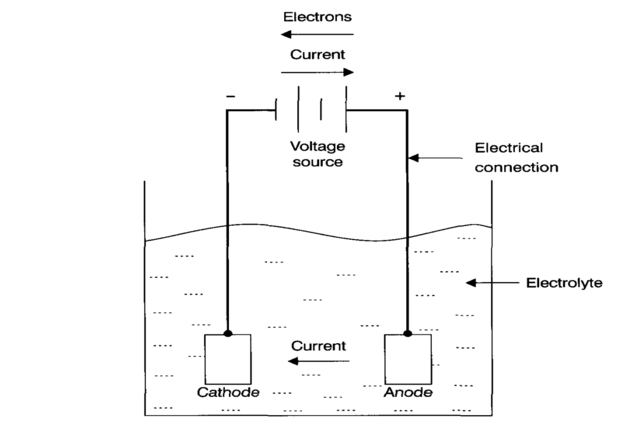
When a metal, such as steel, is placed in an electrolyte (an aqueous solution that can carry ions, like water with some dissolved rock salt), a corrosion cell can form.
Part of the steel in the electrolyte forms the anode, and part in the same electrolyte forms the cathode. Corrosion, in this case, would occur at all anode points distributed throughout the steel (see Figure below). This gives the appearance of general or uniform corrosion.
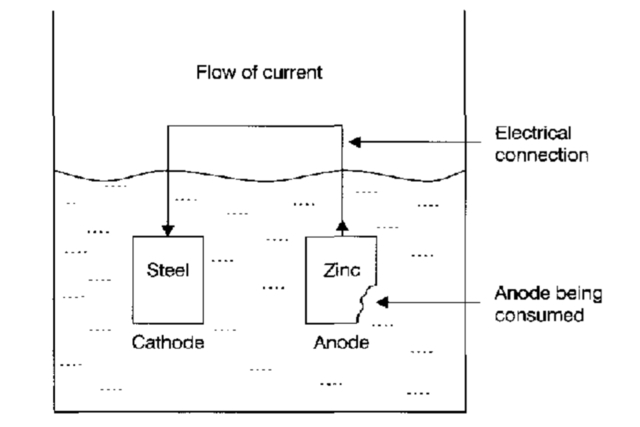
If steel is physically attached (i.e., welded, bolted, or cast) to a piece of zinc and both are immersed in an electrolyte, the zinc will serve as the anode, while the steel will serve as the cathode.
The result will be that all the corrosion reactions will occur on the zinc, which will be consumed, and a balancing reduction reaction (non-corrosion) will occur on the steel, which will not be affected by its immersion in the electrolyte.
This is the basis of cathodic protection. When you cathodically protect steel or any other metal, you change it from acting as either an anode or both an anode and a cathode to acting entirely as a cathode. This is achieved by imposing an external anode that corrodes preferentially (see the Figure below).
Also Read: Engineer | Definition, and History You Should Know Right Now
Corrosion of Steel
In common with all engineering metals, steel is intrinsically unstable in that it wants to return to its stable state, where it came from as an ore.
The result of this reversion is rust (commonly iron oxide, but it can also be iron sulfide or other compounds), which, while having considerably greater chemical stability, also has considerably reduced mechanical properties, such as strength, compared to the original steel.
With this tendency to corrode, the principal question is not whether steel will rust but how fast it will rust.
The environment and the stability of the oxide layer on the surface usually decide the corrosion rate of steel. The reaction rate is slow if this layer forms a protective skin that remains intact.

If, however, the oxide layer is opened at many places and sloughs off the surface, providing access for more oxygen (which is usually dissolved in water) to the unreacted steel surface. A high corrosion rate can be expected.
Straight carbon and high-yield steels are the most commonly used rebar grades in typical civil engineering projects. Neither of these types has a particularly protective oxide film, and both rely on the concrete’s alkalinity to stabilise this skin.
There will be a rapid colour change when steel corrodes in a standard atmosphere, i.e., outdoors. This is known as ‘flash’ resting. For example, blast-cleaned steel in a moist environment changes colour when the contractor finishes the blasting operation and opens the paint pots.
This rusting is evidenced by a change in the surface colour from silver to orangey-red across all exposed surfaces. In this case, the corrosion is very rapid due to ample fuel (oxygen) and the absence of a protective oxide film.
In a saline environment, flash rusting occurs even more quickly, as chloride ions help the water conduct electricity. If the steel were examined visually under a microscope, it would all appear the same colour. In this case, the individual anode and cathode sites are very small, perhaps only a few microns apart.
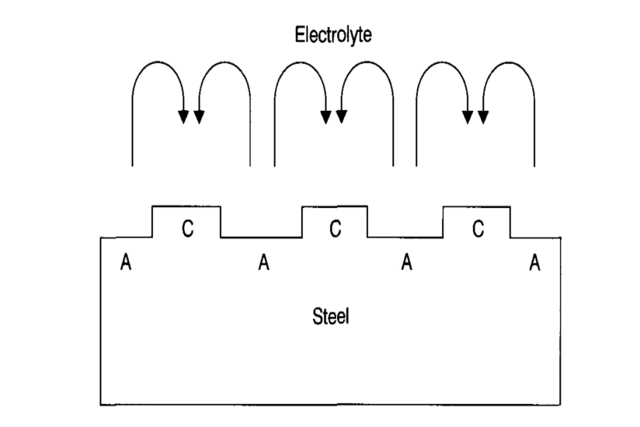
In cases where steel is exposed directly to the atmosphere at a typical (neutral) pH and has a reasonable supply of oxygen, widespread and uniform corrosion will occur.
This is typically observed when extensive steel sections rust and can be seen on any uncovered steel article, particularly on beaches and other areas with a corrosive atmosphere. An example is given in the Figure above.
When oxygen access to the steel is reduced, which becomes the corrosion-limiting step, i.e., when sufficient aggressive ions are present at the steel interface, allowing the corrosion reaction to occur very quickly, other forms of corrosion may occur.
The most common is pitting corrosion. This, for example, occurs when a surface coating on the steel is breached, allowing oxygen and moisture to access a relatively small area. In older cars, these are commonly seen as rust spots. This situation is shown schematically in the Figure below.
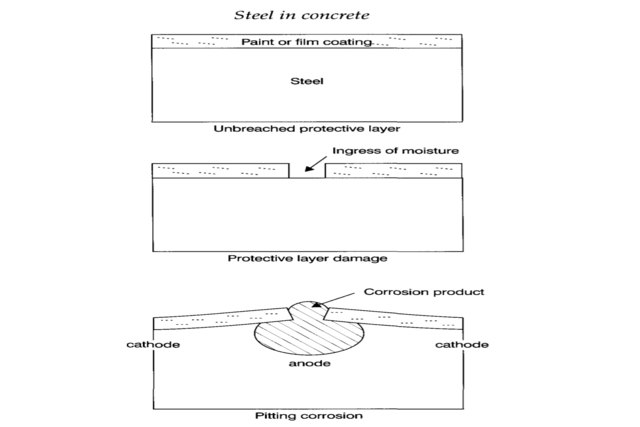
Steel in Concrete
Concrete typically provides embedded steel with a high degree of protection against corrosion. One reason for this is that cement, a constituent of Concrete, is highly alkaline.
This means that the Concrete surrounding the steel provides an alkaline environment for the steel. This stabilises the oxide or hydroxide film, thereby reducing the oxidation (corrosion) rate of the steel.
This state, with a very low corrosion rate, is termed passivation. Another reason Concrete protects embedded steel is that it acts as a barrier against outside elements that are aggressive to the steel.
The chloride ion is the most common agent for the depassivation of steel in Concrete.
The traditional wisdom was that Concrete with a low water-cement ratio and well cured would have a sufficiently low permeability to prevent significant penetration by corrosion-inducing factors, such as oxygen, chloride ions, carbon dioxide, and water. Unfortunately, this is not the proper position.
Some of this can be explained by the fact that Concrete is inherently porous, regardless of its composition. If a concentration gradient exists, then at some point, a sufficient quantity of aggressive ions will be passed through the Concrete to initiate corrosion.
The crux is at ‘some time’ as this might be sufficiently long to achieve the design life, or it might not. Another reason is probably that cracks exist on all full-scale structures, providing preferential pathways for corrosion-inducing factors.
Fortunately, in most steel-reinforced Concrete structures, corrosion does not occur during the design life. However, this is a situation that arises more by luck than judgment.
With Concrete of a suitable quality, corrosion of steel can be prevented for a specific period, provided that the structure or element is correctly designed for the intended environmental exposure.
In severe exposure, such as bridge decks exposed to de-icing salts or pilings in seawater, the permeability of Concrete conforming to specific standards has been measured, and the time required for the onset of corrosion has been calculated using this data, as shown in the Table below.
| Concrete | Minimum cover (mm) | With a chloride concentration of 0.4% | With a chloride concentration of 1% |
| Old pre-cast standard | 38 | 23.7 | 53.8 |
| BS 8110 (general) | 50 | 5.5 | 12.4 |
| BS 5400 (bridges) | 65 | 9.2 | 20.9 |
| BS 6349 (maritime) | 75 | 12.3 | 27.9 |
As can be seen, a corrosion initiation period of only five and a half years is possible when using modern concrete in compliance with the current standard in a particular environment.
Where the Concrete quality and cover of a new structure are insufficient to provide the required design life, several measures may be employed, such as corrosion inhibitors, coatings on the steel and on the Concrete, or cathodic protection.
The correct choice should be determined by evaluating all options for their cost-effectiveness, reliability, and overall effectiveness over the structure’s design life.
If the structure is not corrosion-resistant in the anticipated environment, or if the environment and other factors are not as anticipated or change during the structure’s life, there may be problems.
Instances of distress due to corrosion can be found in almost all applications of modern reinforced or prestressed Concrete; buildings, silos, beams, bridge decks, piles, other supports, tanks, and pipes are some common examples.
Typically, the first indication of distress is the brown staining of the Concrete surface nearest to the corroding embedded steel. This staining is caused by the permeation of iron ions through microcracks in the Concrete to the surface and is often accompanied by the macrocracking of the Concrete shortly afterwards.
The cracking occurs because certain steel corrosion products, such as iron oxides, have a substantially greater volume than the metallic element (iron) from which they were formed. The forces generated by this expansive process exceed the concrete’s tensile strength, resulting in cracking.
Steel corrosion not only causes structural distress or disfigurement due to staining, cracking, and spalling of the Concrete, but also reduces the compressive strength of the structure (due to the damaged Concrete), and more critically, it can often cause structural failure due to the reduced cross-section and hence reduced tensile capacity of the steel.
This reduced tensile capacity is usually significant only in localised areas and is more structurally critical for prestressing steel tendons than for reinforcing bars. This is because the load cannot be easily redistributed in a pre- or post-tensioned structure, unlike in cast-in-situ structures.
As an example of the extreme damage which can occur with steel in cracked and stained Concrete, it is common to find that 60 or 70% of the cross-section has corroded through.
All Concrete corrosion engineers have apocryphal stories about structures where there has been a 100% loss of steel reinforcement section. In certain circumstances, this can be seen at many points.

Mechanisms
When reinforcement steel in Concrete corrodes, the process is similar to taking power from an ordinary battery. When steel corrodes in a battery, a metal dissolves, producing a small current between the positive and negative poles.
In steel reinforcement corroding in concrete, one tiny area is the anode (positive pole), and another, much more extensive area is the cathode (negative pole).
The corrosion current flows out of the steel at the anode, the part that is corroding, through the Concrete and into another part of the steel where no corrosion is occurring, i.e., the cathode. This flow is referred to as a corrosion circuit. Steel dissolves at the anode and eventually forms iron oxide there.
The electrical connection between + and • can be disconnected from a battery. The circuit is then broken, stopping the current and halting the dissolution of the metal.
For steel reinforcement in Concrete, the ionic flow through the Concrete and the attachment between the steel cannot be disrupted, as the corrosion circuit is embedded in the structure.
Instead, it is possible to use an ‘artificial’ anode to add a new, higher current to the original corrosion circuit, which runs in the opposite direction to the corrosion current.
This makes all the previous +poles (anodes) into current receivers. Thus, all the steel reinforcement is made into a negative pole, i.e., cathodic, hence the name ‘cathodic protection.’
In electrochemical corrosion, a flow of electrical current and one or more chemical processes are required for metal loss.
The flow of electrical current can be caused by ‘stray’ electrical sources, such as from a train traction system or from significant differences in potential between parts of the structure caused by factors such as differential aeration from the movement of seawater (the mechanism for this is still uncertain, but it could be that vast cathode areas are built up in the tidal zone because of oxygen charging.
Electrochemical corrosion by these electrical current sources is rare but can be serious when it occurs. Often, this process can contribute to corrosion when other aggressive factors arise.
The passivation of the steel by the alkalinity would likely allow a certain amount of current discharge from the steel without metal loss. The critical factor here is the resupply of alkalinity relative to the current drain.
Instances of stray-current corrosion have been recorded. An example was a jetty where the piles were being cathodically protected, and the reinforced Concrete deck was being used as the system’s negative.
Unfortunately, several of the piles were electrically discontinuous, and corrosion occurred at a secondary anode point formed on these piles as current attempted to flow back to the system in a negative direction.
The vast majority of the potential gradients found between different areas of the steel in Concrete are caused by the existence of physical differences or non-uniformities on the surface of the steel reinforcement (different steels, welds, active sites on the steel surface, oxygen availability, and chloride contamination).
These potential gradients can allow significant electrical current to flow and, under certain circumstances, such as the presence of aggressive ions in the Concrete, cause corrosion of the reinforcement.
Even though electrochemical corrosion may occur due to the non-uniformity of the steel in the Concrete.
This corrosion is usually prevented, even at nominally anodic sites (i.e., more anodic than the cathodic area), by the passivated film that forms on the steel surface in the presence of moisture, oxygen, and water-soluble alkaline products formed during cement hydration.
There are two mechanisms by which the highly alkaline environment and accompanying passivation effect may be destroyed, namely:
- The reduction of alkalinity is achieved by leaching alkaline substances with water or by partial neutralisation when reacting with carbon dioxide or other acidic materials.
- Electrochemical action involves aggressive ions acting as catalysts (typically chloride) in the presence of oxygen.
Reduction of alkalinity by reaction with carbon dioxide, as present either in the air or dissolved in water, involves neutralising reactions with the sodium and potassium hydroxides and, subsequently, the calcium system, which is part of the Concrete matrix.
This process—called carbonation—although progressing increasingly slowly, may in time penetrate the Concrete to a depth of 25mm or so (depending upon the quality of the Concrete and other factors) and thereby neutralise the protective alkalinity normally afforded to steel reinforcement buried to a lesser depth than this.
This damage is particularly apparent in low-grade concrete structures where the builders were economical with the cement and liberal with the water.
The second mechanism by which the passivity of the steel in the concrete can be disrupted is through electrochemical action involving chloride ions and oxygen.
As previously mentioned, this is by far the most critical degradation mechanism for reinforced concrete structures, and the most significant factors influencing this reaction are discussed below.
Also Read: The Contractor | Types, Responsibilities, and Conditions You Need To Know
Alkalinity and chloride concentrations:
The high alkalinity of the chemical environment usually present in Concrete protects the embedded steel because of the formation of a protective film, which could be either an oxide or a hydroxide, or even something in the middle, depending on which research paper you read.
The integrity and protective quality of this film depend on the environment’s alkalinity (pH). The bulk alkalinity of the concrete depends on the water-soluble alkaline products.
The principal soluble product is calcium hydroxide, and the initial alkalinity of the Concrete is at least that of saturated lime water (pH of about 12.4, depending upon the temperature).
Additionally, the cement contains relatively small amounts of sodium and potassium oxides, which can further increase the alkalinity of the Concrete or paste extracts. pH values of 13.2 and higher have been reported.
The higher the alkalinity, the greater the protective quality of this film. Steel in Concrete becomes more susceptible to corrosion as alkalinity decreases.
Additionally, steel in Concrete becomes more susceptible to corrosion as the quantity of soluble chlorides at the iron-cement paste interface increases. Chloride ions appear to be a specific destroyer of the protective oxide film.
Chloride can be present in ‘as-manufactured’ Concrete as a set accelerator (calcium chloride) or through contamination of the mix. However, more commonly, the chloride has come from an external source, such as de-icing salts or marine environments.
In these latter cases, the salt diffuses through the Concrete cover to the steel. It is worth noting that, in practice, the diffusion rate in an exposed marine or de-icing salt environment can be significantly higher than would be estimated from cement or Concrete diffusion tests. This is because the transport process utilises convection and capillary movements in real structures.
Although chloride ions are soluble in the cement paste, most of the chlorides will not be in solution in the liquid within the paste. This is because the chlorides react with hydrated tricalcium sulfoaluminate, a constituent of the past, to produce a corresponding tricalcium chloroaluminate compound.
It has been shown that as much as 75–90% of the chloride in the cement paste exists in the chloroaluminate compound and is thus ‘bound’ and unable to interact with the steel reinforcement.
This concentration depends upon the total amount of chloride present, the tricalcium aluminate (C3A) content, and the degree of hydration of the cement.
Although only a fraction of the chloride is in solution, it is in a dynamic equilibrium. Thus, the chloroaluminate compound present would allow resupply if the free chloride were leached out.
The relationship between the onset of steel corrosion and the alkalinity and chloride concentration in the environment has not been fully defined for in situ concrete.
It is unlikely to be so without a definition of the aggregate and cement types and quantities. It has been suggested, and it seems reasonable, that a threshold concentration of chloride ions must be exceeded in an oxygen-rich environment before significant corrosion of steel reinforcement occurs. Some of the defined values for this level are given in the Table below.
| Authority | % of Cl by weight of cement |
| Wegler | 0.40 |
| BS CP11 1979 | 0.36 |
| Clear | 0.20 |
| Knofel | 0.20 |
| ACI Committee 201 | 0.20 – 0.10 |
| Vassie | 0.10 |
As can be seen, there is an extensive range of defined critical values. Part of the reason is that a scientist would probably define the onset of corrosion as the point at which sufficient chloride is present at the rebar to catalyse the corrosion reaction.
In contrast, if a site investigation reveals a low corrosion rate in commercial structures, then a visual inspection would suggest that no corrosion is occurring.
This observation would allow the erroneous conclusion that the threshold had not been reached. Whatever the critical concentration level, it appears certain that both the structural environment and the Concrete significantly affect the chloride concentration required to initiate corrosion.
A hydroxide-to-chloride concentration ratio can more accurately describe these critical values. This has been given, among others, by Hausmann (1967) as:
It is sometimes found that reinforced Concrete with uniformly high levels of chloride contamination (often over 3%) does not have significant corrosion of the rebar.
This typically occurs when there are constant environmental conditions surrounding the Concrete, such as internal walls of a building or structures buried beneath a saline water table.
Conversely, in areas where there are cyclical environmental conditions, such as where Concrete is exposed to strong tidal flows of aerated salt water or where there are daily weather conditions, e.g., where there is direct sunlight in the day and high humidity and low temperatures in the night, there can be significant damage at low chloride concentrations.
Various ideas have been proposed about how chloride causes this depassivation, but there seems to be agreement that, in localised areas, the passive film is broken down, resulting in pitting.
In the pits, an acidic environment exists, and when Concrete is stripped from corrosion sites on the steel, green-black and yellow-black compounds are often observed.
These are probably intermediate complexes that contain chloride and allow a lower activation energy for oxidation. In the corrosion process, chloride is not held as a final product and can be thought of as acting as a catalyst.
Any increase in chloride ion concentration beyond the initiation level will likely increase the corrosion rate. At some point, other factors will become the rate-limiting step. This rate-limiting step in reinforced Concrete is commonly the availability of sufficient oxygen.
Oxygen level:
A crucial factor in the corrosion of steel in Concrete is the presence of oxygen at the steel-cement paste interface. Oxygen is required in addition to chloride or reduced alkalinity.
If oxygen is not present, there should be no oxidation. For example, seawater has been successfully used as mixing water for reinforced Concrete, where the Concrete is continually and completely submerged in seawater at the seabed.
This is because of the maintenance of high alkalinity due to the sodium chloride (this boosts the Concrete’s alkalinity due to sodium ions’ higher solubility in the cement paste), low oxygen content in the seawater at the seabed, and the very slow diffusion rate of oxygen through the water-saturated paste.
There would initially be a high corrosion rate when the critical chloride ratio was achieved. This would deplete the available oxygen, and the corrosion rate would dramatically reduce, despite an increasing chloride concentration.
This slowing of the corrosion rate is aided by a reduction in the solubility of oxygen in water at extremely high chloride saturation levels, which further reduces oxygen availability.
In most cases, when the structure is submerged, oxygen diffusion is the rate-controlling step in the rate of corrosion.
The level of oxygen supply or resupply also affects the corrosion products formed. A black product (magnetite) is formed in low oxygen availability, and a red-brown material (haematite) is favoured in high oxygen availability.
The pore sizes of these oxides differ, with the red product forming a more open structure featuring larger pores. The formation of haematite imparts a higher bursting pressure on the concrete because of its greater volume.
It allows for a quicker reaction due to its greater porosity relative to magnetite. For these reasons, the presence of haematite rather than magnetite tends to indicate general corrosion rather than pitting and vice versa.
Cement type:
The Concrete composition significantly affects corrosion damage at varying chloride concentrations. One example is that hardened Concrete appears to have a lower chloride tolerance than concrete contaminated during mixing.
This is practically evident in pre-cast units, which tend to corrode less than might be anticipated, even when heavily dosed with a calcium chloride set accelerator (this is probably at least partially explainable due to their higher quality relative to cast-in-situ reinforced concrete of the same vintage and the absence of any potential differences caused by concentration gradients).
Although the cement composition and type can affect corrosion, this effect is relatively small compared to the quality of the concrete, the cover over the steel, and the consolidation of the Concrete.
Using cement with a high C3A content tends to bind more chlorides, thereby reducing the amount of chlorides available to disrupt the oxide film on the steel reinforcement.
Additionally, a cement with a high alkali content would appear to offer advantages due to its higher inherent alkalinity. In general, it is observed that cement high in C3A affords better corrosion protection to reinforcing steel. Still, it is thought that other factors, such as fineness and sulfate content, may have at least as significant an effect.
One study by Tuutti (1982) found that Portland cement had a higher initiation level than slag cement. Still, lower diffusion resistance is postulated, suggesting that under certain exposure conditions, a specific Portland cement mix design would be superior. In contrast, under other conditions, the reverse would hold: slag cement would be exceptional.
It was noted that sulfate-resisting cement was always less effective than Portland cement.
Aggregate type and other additives:
In general, the higher the strength of the aggregate, the more likely it is to be resistant to the passage of ions. But this is not always so.
For example, granite aggregate has been used in several significant projects due to its high strength; however, Concrete made with this material has been found to exhibit relatively poor diffusion resistance. This is likely due to microcracks in the aggregate.
Likely, a substantial amount of the diffusion in Concrete proceeds along the interface between the aggregate and the cement paste. This region may prove more critical than the aggregate’s bulk diffusion resistance. Some aggregates have a smoother profile than others, which affects the apparent diffusion path.
Adding additives, such as microsilica, to concrete is beneficial, as it increases the diffusion path by blocking pores in the Concrete.
The primary concern with this and other additives is the additional care required when casting on the construction site, as well as the assumption that the Concrete will become impermeable with additive X. This is not a safe assumption, as the Concrete will still retain some degree of porosity.
Join the conversation by replying on Bluesky.
That’s all.

")